Endoplasmic reticulum (ER) Ca2+-channel activity contributes to ER stress and cone death in cyclic nucleotide-gated channel deficiency
11-May-2017
J. Biol. Chem., 292(27) 11189–11205, DOI 10.1074/jbc.M117.782326
J. Biol. Chem., online article
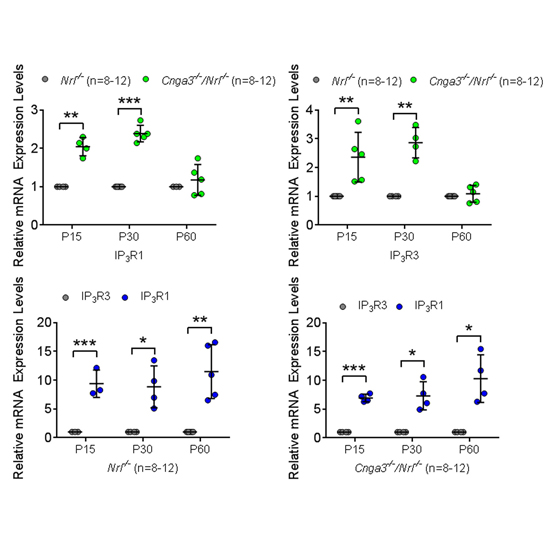
Endoplasmic reticulum (ER) stress and mislocalization of improperly folded proteins have been shown to contribute to photoreceptor death in models of inherited retinal degenerative diseases. In particular, mice with cone cyclic nucleotide-gated (CNG) channel deficiency, a model for achromatopsia, display both early-onset ER stress and opsin mistrafficking. By 2 weeks, these mice show elevated signaling from all three arms of the ER stress pathway, and by 1 month, cone opsin is improperly distributed away from its normal outer segment location to other retinal layers. This work investigated the role of Ca2+ release channels in ER stress, protein mislocalization, and cone death in a mouse model of CNG channel deficiency. We examined whether preservation of luminal Ca2+ stores through pharmacological and genetic suppression of ER Ca2+ efflux protects cones by attenuating ER stress. We demonstrated that inhibition of ER Ca2+ efflux channels reduced all three arms of ER stress signaling while improving opsin trafficking to cone outer segments and decreasing cone death by 20-35%. Cone-specific gene deletion of the inositol-1,4,5-trisphosphate receptor type I (IP3R1) also significantly increased cone density in the CNG channel-deficient mice, suggesting that IP3R1 signaling contributes to Ca2+ homeostasis and cone survival. Consistent with the important contribution of organellar Ca2+ signaling in this achromatopsia mouse model, significant differences in dynamic intraorganellar Ca2+ levels were detected in CNG channel-deficient cones. These results thus identify a novel molecular link between Ca2+ homeostasis and cone degeneration; thereby revealing novel therapeutic targets to preserve cones in inherited retinal degenerative diseases.