Mosaic synaptopathy and functional defects in Cav1.4 heterozygous mice and human carriers of CSNB2
26-Oct-2013
Hum. Mol. Genet., 2013, doi: 10.1093/hmg/ddt541, published on 26.10.2013
Hum. Mol. Genet., online article
Hum. Mol. Genet., online article
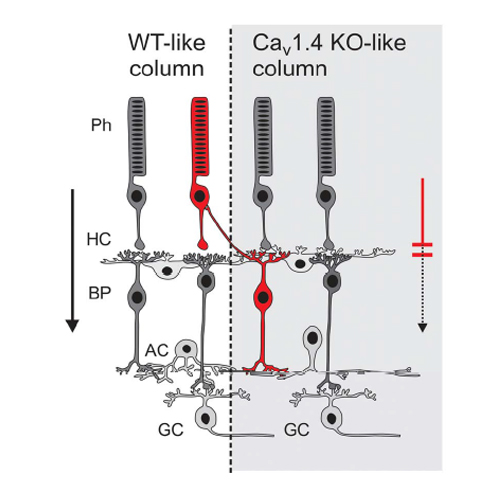
Mutations in CACNA1F encoding the α1-subunit of the retinal Cav1.4 L-type calcium channel have been linked to Cav1.4 channelopathies including incomplete congenital stationary night blindness type 2A (CSNB2), Åland Island eye disease (AIED) and cone-rod dystrophy type 3 (CORDX3). Since CACNA1F is located on the X chromosome, Cav1.4 channelopathies are typically affecting male patients via X-chromosomal recessive inheritance. Occasionally, clinical symptoms have been observed in female carriers, too. It is currently unknown how these mutations lead to symptoms in carriers and how the retinal network in these females is affected. To investigate these clinically important issues, we compared retinal phenotypes in Cav1.4-deficient and Cav1.4 heterozygous mice and in human female carrier patients. Heterozygous Cacna1f carrier mice have a retinal mosaic consistent with differential X-chromosomal inactivation, characterized by adjacent vertical columns of affected and non-affected wild-type-like retinal network. Vertical columns in heterozygous mice are well comparable to either the wild-type retinal network of normal mice or to the retina of homozygous mice. Affected retinal columns display pronounced rod and cone photoreceptor synaptopathy and cone degeneration. These changes lead to vastly impaired vision-guided navigation under dark and normal light conditions and reduced retinal electroretinography (ERG) responses in Cacna1f carrier mice. Similar abnormal ERG responses were found in five human CACNA1F carriers, four of which had novel mutations. In conclusion, our data on Cav1.4 deficient mice and human female carriers of mutations in CACNA1F are consistent with a phenotype of mosaic CSNB2.